Wilson’s disease isn’t something you hear about often, but if you or someone you know has unexplained liver problems, tremors, or trouble speaking, it could be the hidden cause. This rare genetic disorder doesn’t just affect the liver-it quietly floods the body with toxic copper until organs start failing. The good news? If caught early, it’s treatable. The bad news? Most people go years without a correct diagnosis.
How Copper Turns Toxic in Wilson’s Disease
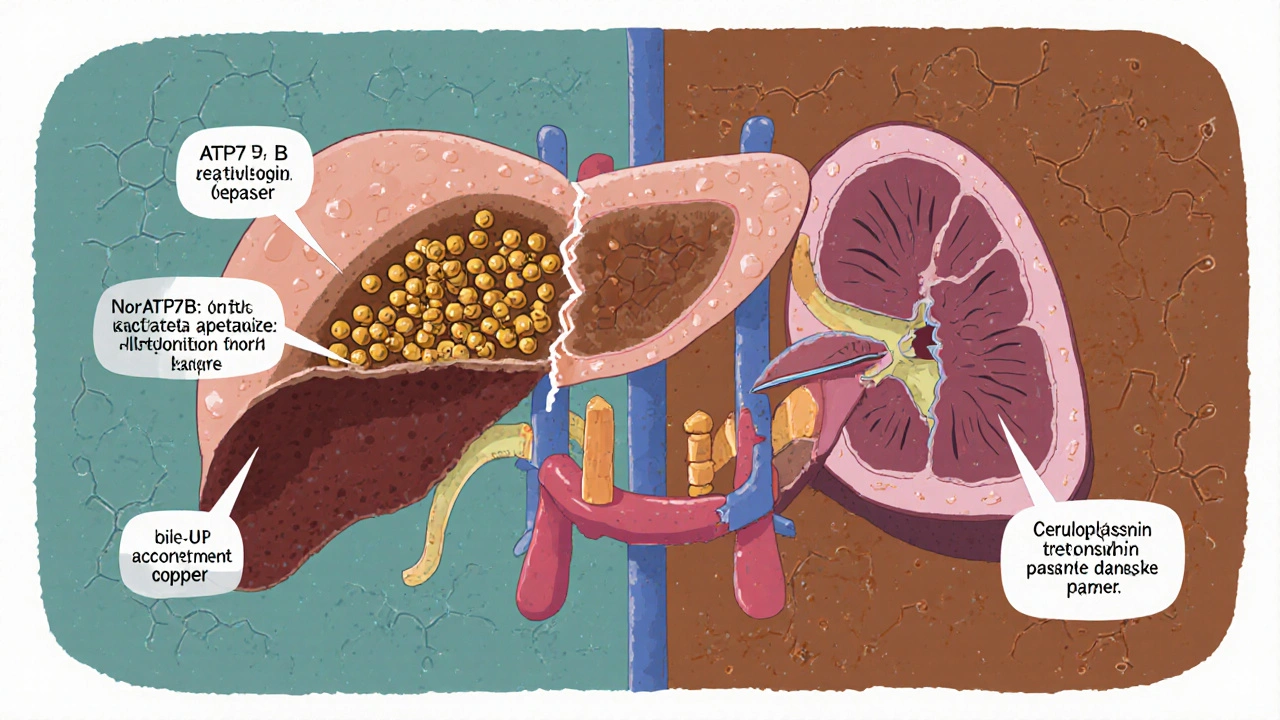
Your body needs copper. It’s in your blood, your brain, your bones. But too much? It’s poison. In Wilson’s disease, a broken gene called ATP7B stops the liver from doing its job: getting rid of extra copper. Normally, the liver packages copper into a protein called ceruloplasmin or pushes it out through bile into the gut. In Wilson’s disease, that process breaks down.
Instead of leaving the body, copper builds up in the liver. At first, it’s stored safely by proteins like metallothionein. But once those storage tanks are full, copper spills into the bloodstream as free copper-unattached, reactive, and dangerous. This is what damages the brain, kidneys, and eyes.
The telltale sign? Kayser-Fleischer rings. These rusty-brown rings around the cornea aren’t visible to the naked eye unless you’re examined with a slit-lamp. They appear in 95% of people with neurological symptoms and are a major red flag for doctors. But here’s the catch: in kids under five, these rings often don’t show up yet. That’s why many children are misdiagnosed with hepatitis or fatty liver disease.
Why Diagnosis Is So Hard
Wilson’s disease mimics other conditions. Liver enzymes go up? Autoimmune hepatitis. Tremors and stiff movements? Parkinson’s. Depression and personality changes? Mental health issue. That’s why the average time to diagnosis is nearly three years. One patient on Reddit shared: "I had abnormal liver tests for seven years. Two doctors told me it was stress. Then my urine copper came back at 380 μg/24h-and everything clicked."
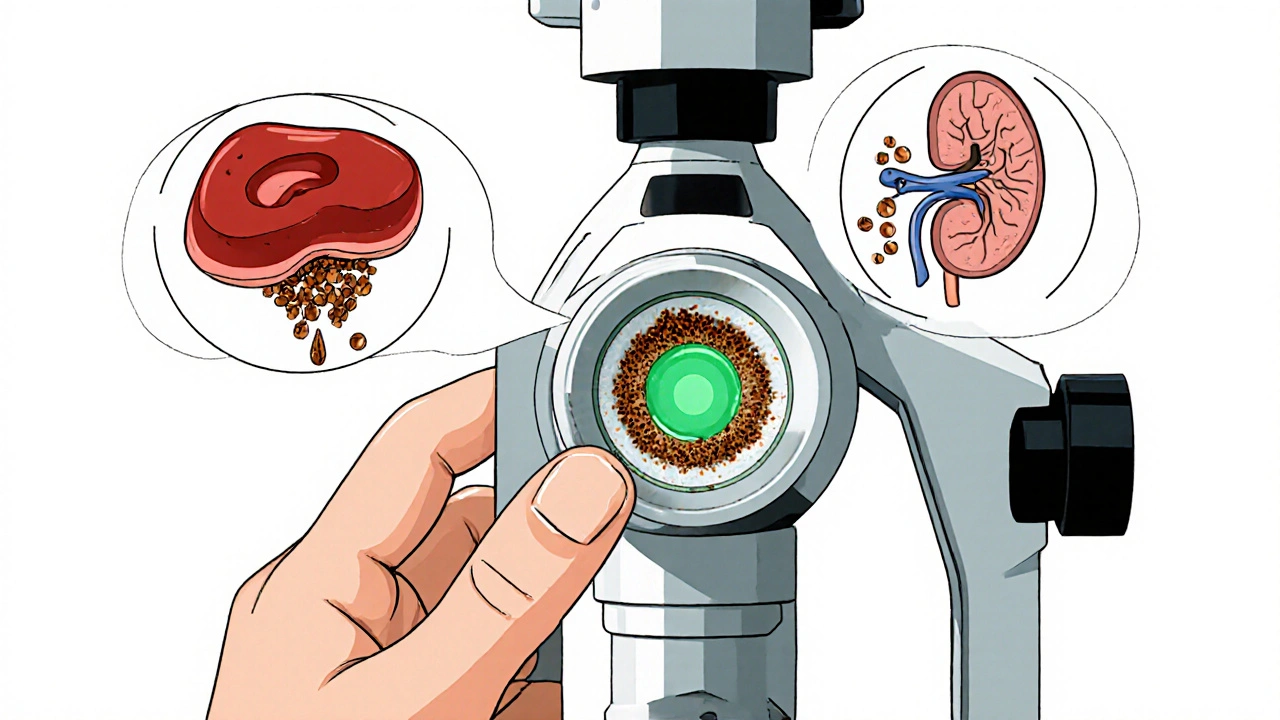
Standard tests include:
- Serum ceruloplasmin (usually below 20 mg/dL in WD, normal is 20-50)
- 24-hour urinary copper (over 100 μg/24h is highly suggestive)
- Serum free copper (above 25 μg/dL indicates toxicity)
- Slit-lamp eye exam for Kayser-Fleischer rings
- ATP7B gene testing (definitive confirmation)
But here’s the twist: some patients with neurological symptoms have normal or only slightly low ceruloplasmin. Others with liver disease have urinary copper below 100 μg/24h. That’s why experts now recommend using a scoring system that combines all these factors-and why genetic testing is becoming the gold standard.
Chelation Therapy: The Lifesaving Fix
Once diagnosed, treatment starts immediately. The goal? Pull copper out of the body without making things worse. That’s where chelation therapy comes in.
The most common first-line drug is D-penicillamine. It binds copper and lets the kidneys flush it out. But it’s a double-edged sword. About 30% of patients get worse in the first few weeks-tremors spike, speech gets slurred, movement becomes harder. Why? Because as copper is pulled from tissues, it temporarily floods the brain before being cleared.
To prevent this, doctors often add zinc alongside penicillamine. Zinc doesn’t remove copper-it blocks it. It triggers the gut to make metallothionein, a protein that traps copper from food so it can’t be absorbed. That’s why patients on zinc are told to take it on an empty stomach-food interferes.
Another option is trientine. It’s gentler on the nervous system and causes fewer neurological side effects, but it costs over $1,800 a month in the U.S. Penicillamine? Around $300. Cost matters, especially when treatment lasts a lifetime.
Then there’s zinc acetate. It’s not a chelator-it’s a maintenance drug. Once copper levels drop, patients often switch to zinc long-term. It’s safer, cheaper, and prevents copper from building up again. Studies show 92% of patients on zinc stay stable if their free copper stays under 10 μg/dL.
What Patients Actually Experience
Living with Wilson’s disease isn’t just about taking pills. It’s about diet, side effects, and constant monitoring.
Patients are told to avoid high-copper foods: shellfish, liver, mushrooms, nuts, chocolate, and even some tap water (if pipes are copper). The daily limit? 1 mg. Sounds simple? Try it. One patient said, "I gave up my morning oatmeal with almonds. My lunch was a salad with no spinach. Dinner? Plain chicken. I missed everything."
Side effects are common. Nearly 75% of patients report them:
- Penicillamine: nausea, metallic taste, skin rashes, lupus-like symptoms
- Trientine: iron deficiency (35% of users), stomach pain
- Zinc: low copper levels if overused, anemia
And adherence? Only 65% take their meds as prescribed. Missing a dose can mean copper creeps back up. One woman on a support forum wrote: "I skipped my zinc for two days because I was traveling. My ALT jumped from 40 to 110 in a week. I learned the hard way: no exceptions."
New Treatments on the Horizon
There’s hope beyond the old drugs. In 2023, a new copper-binding polymer called CLN-1357 showed an 82% drop in free copper in just 12 weeks-with no neurological worsening. That’s huge. No more initial spikes in tremors.
Another promising drug, WTX101 (bis-choline tetrathiomolybdate), got breakthrough status from the FDA in early 2023. In trials, it prevented neurological decline in 91% of patients, beating trientine’s 72%. It also crosses the blood-brain barrier better, meaning it can reach brain copper faster.
And then there’s gene therapy. Early trials are injecting a working copy of the ATP7B gene into the liver using harmless viruses. Six patients so far have shown no serious side effects. It’s not a cure yet, but it’s the first real shot at fixing the root cause-not just managing symptoms.

What Happens If You Don’t Treat It
Left untreated, Wilson’s disease kills. Liver failure. Brain damage. Kidney damage. The copper doesn’t just sit there-it causes oxidative stress, destroys cells, and triggers inflammation. Without chelation, most people die by age 30.
But with treatment? Life expectancy becomes normal. A 2022 study tracked 400 patients over 20 years. Those who stuck to their meds and monitoring lived just as long as people without the disease. The key? Consistency. Not perfection. Not being flawless. Just showing up.
Final Thoughts: It’s Manageable, But Never Ignorable
Wilson’s disease isn’t common. But it’s deadly if missed. If you have unexplained liver issues, neurological symptoms, or a family history of early liver disease or psychiatric disorders, push for testing. Don’t wait for Kayser-Fleischer rings. Don’t assume it’s stress or anxiety. Ask for a 24-hour urine copper test. Ask for ceruloplasmin. Ask for gene testing.
Treatment isn’t easy. It’s expensive. It’s complicated. But it works. Thousands of people are alive today because they got diagnosed-and stuck with the plan. The future holds better drugs, maybe even a cure. But right now, the best weapon is awareness. Know the signs. Ask the questions. Don’t let copper go unnoticed.
13 Comments
swatantra kumar
Copper poisoning like this is wild 😅 I thought only old people got liver issues from booze, not from their own genes. My uncle had tremors for years and they called it 'stress'-turns out he was basically a walking toxic waste dump. Glad we can fix it now. 🙌
Cinkoon Marketing
I work in a clinic and we see this way more than people think. Most docs just throw a liver panel at you and say 'it's fatty liver' and send you on your way. The fact that ceruloplasmin can be normal and you still have WD? That's the trap. I always push for urinary copper if there's any neuro symptoms. Even if it's 'just anxiety'.
Matthew McCraney
Y'all ever think this is all a Big Pharma scam? Copper is essential, right? So why are they pushing expensive chelators? What if the real problem is glyphosate in our food? Or 5G messing with metallothionein? I read a paper on a forum where a guy cured his WD with apple cider vinegar and a Himalayan salt bath. They silenced him. I know it.
serge jane
The real tragedy here isn't the disease it's how we treat illness as a checklist instead of a story. We reduce a human being to ceruloplasmin levels and urinary copper and gene markers but forget that the person is the one living with the nausea the metallic taste the fear of forgetting to take zinc on an empty stomach while traveling. Treatment isn't just chemistry it's endurance. And endurance isn't measured in labs it's measured in mornings you still get up and swallow the pill even when you hate it.
Nick Naylor
Let me be clear: this is a genetic disorder that disproportionately affects certain populations. The fact that we're even discussing 'chelation therapy' as if it's some new miracle is absurd. We've known about ATP7B since the 90s. Why is this still a problem in the U.S.? Because of healthcare fragmentation. Insurance won't cover trientine. Doctors won't test unless it's 'classic'. We need a national screening protocol. Not a blog post. A law.
Brianna Groleau
I had a friend who was diagnosed at 19. She gave up chocolate. She gave up almonds. She gave up her favorite mushroom risotto. She cried in the grocery store because she couldn't find a single safe snack. But now? She's a nurse. She works in neurology. She tells every patient with unexplained tremors: 'Ask for the copper test.' She turned her pain into purpose. That's the real win. Not the drug. Not the gene. The person.
Rusty Thomas
I saw a TikTok of a guy with WD doing a dance challenge and he had the BEST tremors ever 😭 I mean like he was literally popping and locking with the shakes. I thought it was performance art. Turns out he's been on zinc for 8 years. Now he's doing stand-up comedy about it. I'm not crying you're crying. 🥲
Sarah Swiatek
Funny how penicillamine makes you worse before you get better. Like life. You have to go through the storm to get to the calm. I’ve been on trientine for 12 years. I still get that metallic taste every morning. I still check my urine copper every month. I still hate the diet. But I’m alive. My daughter’s 7. She just got her first gene test. Negative. I cried for an hour. Not because I’m relieved. Because I finally get to be a mom without a countdown clock ticking.
Dave Wooldridge
They’re hiding the truth. Copper doesn’t just accumulate. It’s being injected. Into the water supply. Into the vaccines. Into the 5G towers. They don’t want you to know that Wilson’s Disease is a manufactured crisis to sell chelators. Look at the patents. Look at the funding. The FDA is owned by the same people who own the drug companies. I’ve got screenshots. I’ve got sources. They’re coming for you next.
rob lafata
You people are so naive. You think this is about copper? Nah. It’s about control. They want you dependent. They want you taking pills every day. They want you scared of nuts and chocolate. They want you paying $1800 a month for trientine so you never question the system. You’re not sick. You’re programmed. Wake up. Stop buying into the copper narrative. Go off meds. Eat liver. Let the copper flow. It’s your body’s wisdom.
Rebecca Cosenza
I skipped my zinc for two days. My ALT jumped. Don’t be me. 🙏
robert cardy solano
I used to work in a lab that ran copper tests. We’d see the same names over and over. Same patients. Same numbers. Same stories. One guy came in every month for 12 years. Never missed a test. Never complained. Just smiled and said 'still here.' I asked him why. He said: 'Because I get to watch my granddaughter grow up.' That’s the real treatment. Not the pill. The love.
Pawan Jamwal
In India we don’t even test for this. People die from liver failure and no one knows why. My cousin’s son died at 14. They said 'viral hepatitis.' Now I know. If you’re in India and have unexplained tremors or liver issues - demand the copper test. Don’t wait for a doctor to think of it. Fight for it. We can’t wait for Western medicine to save us.